
La neuromyélite
optique
La maladie, description, symptômes et traitements
INTRODUCTION
La Neuromyélite optique s’exprime par une inflammation du système nerveux central touchant principalement la moelle épinière* et le nerf optique*. Anciennement appelée «maladie de Devic », la NMO regroupe un ensemble de pathologies auto-immunes rares qui se manifestent dans plus de 90 % des cas par des poussées imprévisibles.
Une poussée correspond à un nouveau symptôme ou l’aggravation d’un symptôme existant s’installant de façon rapide, quelques heures à quelques jours. Il existe actuellement deux types de Neuromyélite optique dans lesquelles sont retrouvés des anticorps* : La NMO avec présence d’anticorps anti-AQP4* (NMO AQP4+) et la NMO avec présence d’anticorps anti-MOG (MOGAD*). Dans un faible pourcentage de NMO, aucun anticorps n’est retrouvé. La NMO n’est pas d’origine génétique, ce n’est pas une maladie transmissible, ni une infection.
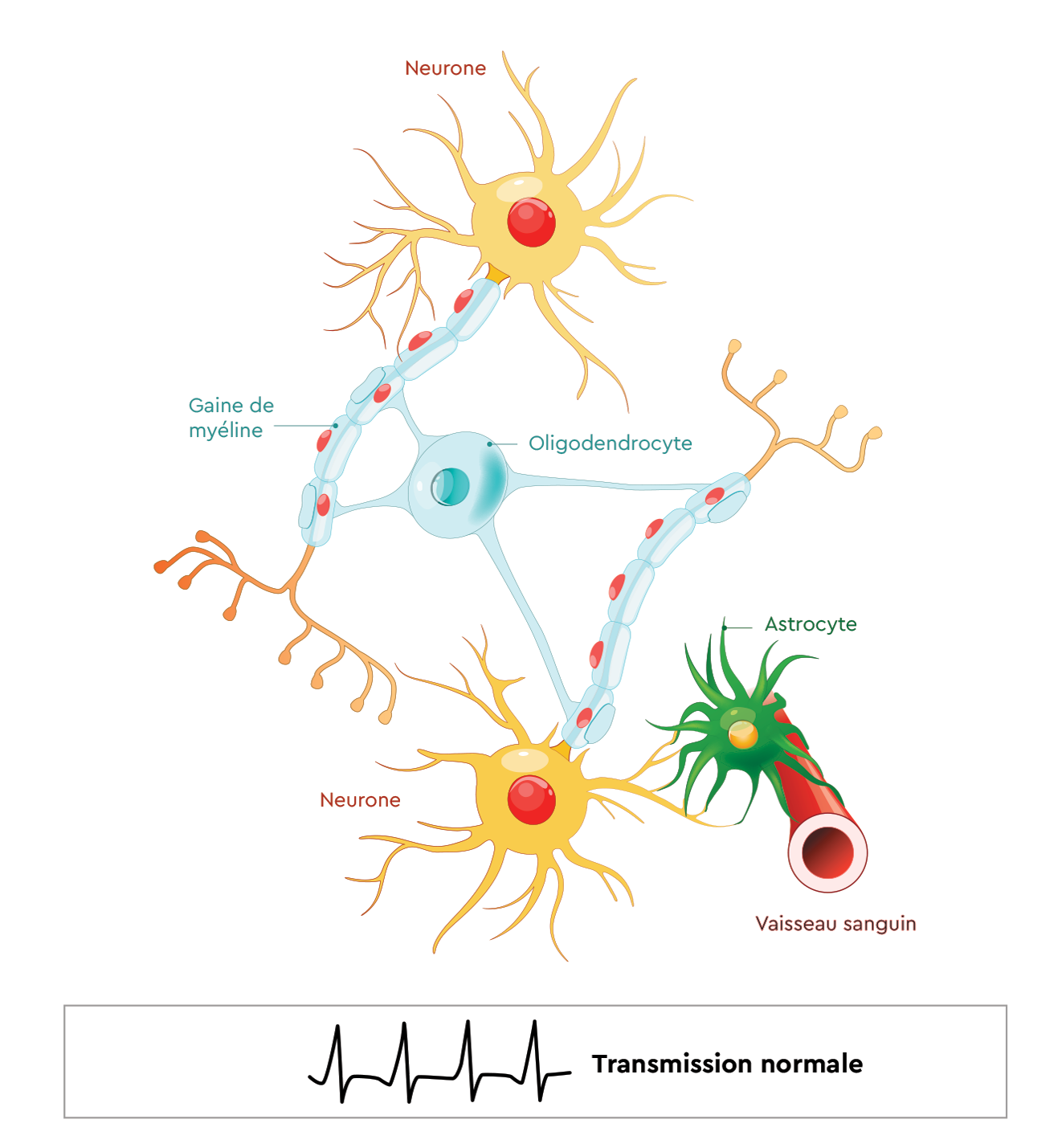
Comment fonctionne le SNC ?
- Le système nerveux central est composé de cellules appelées neurones*.
- Les neurones permettent la transmission de l’influx nerveux (ordre envoyé du SNC vers l’organisme) sous forme de signaux électriques.
- Pour assurer une bonne transmission de ces signaux, le neurone va être enveloppé d’une gaine appelée gaine de myéline*. Cette dernière est produite par une cellule appelée oligodendrocyte*.
- Pour fonctionner, ces cellules ont besoin d’être « nourries ». C’est le rôle joué par l’astrocyte*.
- Cette cellule est en contact avec les vaisseaux sanguins d’un côté et avec les cellules du système nerveux central de l’autre, permettant ainsi de capter les éléments du sang pour les apporter aux neurones et oligodendrocytes.
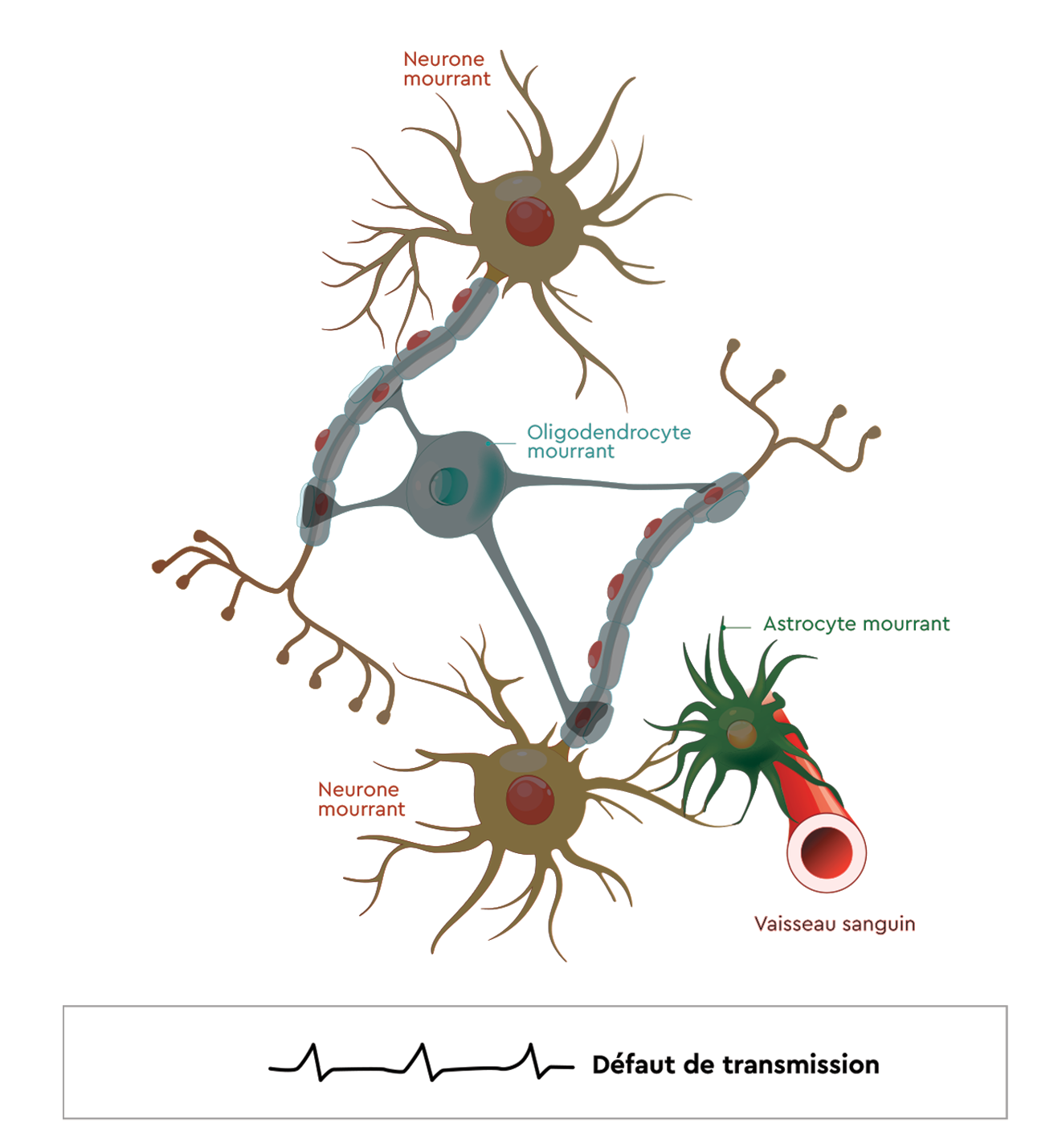
Que se passe-t-il dans le SNC
en cas de NMO ?
En cas de NMO, l’organisme va produire des anticorps qui vont cibler des éléments sur des cellules saines : astrocytes, oligodendrocytes.
Lorsque l’anticorps se fixe sur ces cellules, il va activer le système immunitaire.
Ce dernier va attaquer ces cellules et les détruire.
Conséquence :
• Destruction des astrocytes
• Destruction de la gaine de myéline
• Mort neuronale
En résulte un défaut de transmission de l’information envoyée par le SNC vers le reste de l’organisme, responsable des symptômes de la NMO.
Crédit image : Alexion
-
EPIDEMIOLOGIE
La « neuromyélite optique aigüe » a été décrite pour la première fois en 1894. Elle est connue sous le nom de maladie de Devic depuis 1907, mais ce groupe de maladie étant assez rare, ce n’est qu’en 1999 que les premiers critères diagnostics (Mayo Clinic) ont été publiés.
La terminologie initiale a été remplacée par le terme « NMOSD » pour NeuroMyelitis Optica Spectrum Disorders, maladies du spectre de la neuromyélite optique. De nouveaux critères diagnostics ont été publiés en 2006 et révisés en 2015 (critères de Wingerschuck).
Ces maladies peuvent toucher les enfants et survenir après 70 ans. L’âge moyen en début de maladie est de 40ans.
Dans la NMO à anticorps anti aquaporine 4 (AQP4+), on compte 7 à 9 femmes pour 1 hommes atteint, alors que le nombre d’hommes et de femmes est sensiblement le même en cas d’anticorps anti MOG (MOGAD = Myelin Oligodendrocyte Glycoprotein antibody Associated Disease). Les MOGAD survenant chez l’enfant touchent autant les filles que les garçons.
Les NMO AQP4+ représentent 73 à 90 % des NMOSD, les MOGAD, 5 à 25%.
Les NMO AQP4+ peuvent toucher des non-caucasiens. 30% des patients ont une autre maladie auto immune (myasthénie, Lupus, thyroïdites, etc.)
-
PHYSIOPATHOLOGIE
Les anticorps, anti AQP4 et anti MOG sont responsables d’une attaque de certains composants du système nerveux qui aboutit à une inflammation qui entraîne une atteinte de la gaine de myéline.
La détérioration de cette gaine ne permet plus une transmission normale de l’influx nerveux : les informations sont conduites imparfaitement et/ou plus lentement ou non transmises. Cela se traduit par de symptômes neurologiques : troubles visuels, sensitifs, fatigabilité musculaire, troubles urinaires, etc. Ces symptômes varient en fonction de l’endroit où se développe l’inflammation: dans la moelle, dans le nerf optique ou le cerveau
Ces maladies ne sont pas d’origine génétique, elles ne sont pas transmissibles comme une maladie infectieuse.
-
CLINIQUE
Plusieurs symptômes neurologiques peuvent survenir :
- La névrite optique
La névrite optique correspond à une inflammation d’une partie d’un ou des deux nerfs optiques.
C’est le premier symptôme de la maladie dans plus de 50% des cas.
Cette inflammation entraîne l’apparition d’une vision floue, s’installant généralement en quelques heures, touchant généralement un œil, mais pouvant atteindre les deux, dont l’importance est variable d’une personne à l’autre. Elle peut s’accompagner d’une douleur située le plus souvent derrière l’œil, lors des mouvements des yeux. Elle est fréquemment associée à une difficulté à distinguer certaines couleurs et à une amputation du Champ visuel.
La Récupération est plus ou moins rapide, en fonction de l’efficacité du traitement réalisé.
- La myélite transverse
Une myélite correspond à une inflammation plus ou moins étendue de la moelle épinière.
Il s’agit de la première manifestation de la maladie dans 30 à 40 % des cas.
Elle est le plus souvent d’apparition progressive et se traduit par plusieurs symptômes qui peuvent être isolés ou associés, comme des troubles de la sensibilité, des troubles de l’équilibre, des difficultés à la marche ou des difficultés à uriner avec besoin plus fréquent, difficultés à vider entièrement sa vessie, impression de mal la vider, avec parfois une constipation ou des diarrhées et des difficultés sexuelles (troubles vésicosphinctériens).
Ces symptômes peuvent toucher une ou deux jambes et/ou un ou deux bras.
- Les autres symptomes
Les personnes atteintes de neuromyélite optique peuvent avoir des vomissements, un hoquet ou un prurit (besoin de se gratter), d’origine neurologique ou encore une vision double, des troubles de la sensibilité ou de la motricité d’une moitié du visage, une modification de la voix ou faire des fausses routes, bien que cela soit plus rare.
-
DIAGNOSTICS
Le diagnostic repose aujourd’hui sur trois éléments principaux :
Ce que l’on appelle l’histoire clinique : les symptômes présentés et leur évolution
L’IRM (cérébrale et médullaire) sur lesquelles ont recherche des inflammations
Le bilan sanguin et la ponction lombaire, nécessaires pour affirmer le diagnostic et éliminer d’autres diagnostics (maladie de Lyme, sclérose en plaques, autres maladies neurologiques inflammatoires). Ils permettent de rechercher des Anticorps spécifiques de la NMO : les Anticorps anti Aquaporine 4 et les anticorps anti MOG
-
EVOLUTION
Les MOGAD évoluent par poussée. La poussée correspond à une phase aiguë de la maladie ou de nouveaux symptômes neurologiques apparaissent.
Chaque poussée est différente selon l’endroit de l’inflammation et en fonction des individus.
Sa prise en charge est une urgence! De la rapidité du traitement va dépendre la récupération
En l’absence de traitement les poussées surviennent au rythme d’une par an.
- La NMO n’évolue pas entre les poussées
-
TRAITEMENTS
Le traitement des poussées
Une poussée doit être traitée rapidement par injections intraveineuses de 1 g de CORTICOÏDES. En fonction de la sévérité des symptômes, 3 ou 5 perfusions, ou plus, sont organisées.
En fonction de l’évolution, il peut être nécessaire d’avoir recours à des échanges plasmatiques, traitement hospitalier qui consiste à filtrer le sang pour enlever les anticorps responsables de l’inflammation.
Les traitements de fond
Ce traitement est indispensable dans la NMO AQP4 pour éviter la survenue de nouvelles poussées et l’installation d’un handicap. Il existe plusieurs traitements qui interviennent tous en évitant la réaction immunitaire responsable de la maladie et intervenant à différents niveaux de cette réaction immunitaire.
Dans le cadre d’une MOGAD, le traitement sera discuté en fonction des différents éléments clinique et biologiques.
Les traitements symptomatiques
Ils permettent de soulager les divers symptômes liés à la maladie.
La prise en charge en kinésithérapie et la rééducation favorisent la récupération et aident à compenser d’éventuelles séquelles.
Le traitement des symptômes douloureux peuvent être proposés.
Les troubles sphinctériens et sexuels peuvent faire l’objet d’une prise en charge par des spécialistes ; médecins réducteurs, urologues, gynécologues car de nombreux traitements et prise ne charge existent.
Une prise en charge psychologique est parfois nécessaire pour apprendre à vivre avec une maladie chronique.
-
VACCINS
Il est important que les vaccinations obligatoires soient à jour. Il n’y a pas de contre-indication à se faire vacciner.
Avec certains traitements, certaines vaccinations sont obligatoires et d’autres sont contre-indiquées :
Posez la question à votre neurologue !
-
GROSSESSE
Dans les NMOSD, la grossesse n’est pas contre indiquée, mais il est souhaitable d’évoquer ce sujet avec votre neurologue afin de permettre de prendre les bonnes décisions pour l’envisager sereinement.
VIE QUOTIDIENNE
- L’alimentation doit être variée et équilibrée. Il n’existe pas de régime conseillé.
- Un sommeil régulier, une activité physique, voire sportive hebdomadaire, sont vivement encouragés.
- Aucun voyage n’est interdit, cependant, il est souhaitable de faire le point sur ses vaccinations avant de partir.
- Il est souhaitable de poursuivre son activité professionnelle, dans la mesure du possible et avec l’aide, si nécessaire du médecin du travail.
- Les médecines complémentaires (hypnose, acupuncture, sophrologie, etc.) peuvent aider dans la gestion du stress, de la fatigue, des douleurs, des problèmes de sommeil, etc.
ALSACEP
Hôpitaux Civils de Colmar
Bat 9B – Rez-de-Chaussée
39 avenue de la Liberté - 68024 COLMAR cedex
03.89.30.54.17
infos@alsacep.org
Imaginé et réalisé par : Agence LACOM
Site réalisé avec le soutien institutionnel de
Le réseau ALSACEP fait partie de la Fédération neurologique du Grand Est
